Reference-free cell-type deconvolution of multi-cellular spatially resolved transcriptomics data
![]()
STdeconvolve enables reference-free cell-type deconvolution of multi-cellular pixel-resolution spatial transcriptomics data. The overall approach is detailed in the official paper out in Nature Communications.
Overview
STdeconvolve is an unsupervised machine learning approach to deconvolve multi-cellular pixel-resolution spatial transcriptomics datasets in order to recover the putative transcriptomic profiles of cell-types and their proportional representation within spatially resolved pixels without reliance on external single-cell transcriptomics references.

Installation
To install STdeconvolve, we recommend using remotes:
require(remotes)
remotes::install_github('JEFworks-Lab/STdeconvolve')
STdeconvolve is also now available through Bioconductor.
Note that through Bioconductor (release 3.15), the R version must be >=4.2.
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
# The following initializes usage of Bioc devel
BiocManager::install(version='devel')
BiocManager::install("STdeconvolve")
Installation should take a few minutes on a typical desktop computer.
Notes about package branches
The default package branch R dependency is >=4.1, however, the devel branch is >=3.6.
Example
library(STdeconvolve)
## load built in data
data(mOB)
pos <- mOB$pos
cd <- mOB$counts
annot <- mOB$annot
## remove pixels with too few genes
counts <- cleanCounts(cd, min.lib.size = 100)
## feature select for genes
corpus <- restrictCorpus(counts, removeAbove=1.0, removeBelow = 0.05)
## choose optimal number of cell-types
ldas <- fitLDA(t(as.matrix(corpus)), Ks = seq(2, 9, by = 1))
## get best model results
optLDA <- optimalModel(models = ldas, opt = "min")
## extract deconvolved cell-type proportions (theta) and transcriptional profiles (beta)
results <- getBetaTheta(optLDA, perc.filt = 0.05, betaScale = 1000)
deconProp <- results$theta
deconGexp <- results$beta
## visualize deconvolved cell-type proportions
vizAllTopics(deconProp, pos,
groups = annot,
group_cols = rainbow(length(levels(annot))),
r=0.4)
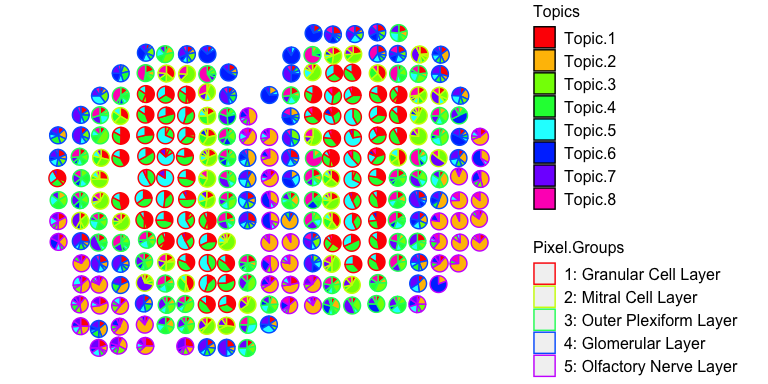
More details can be found in the tutorials.
Tutorials
- Getting started with
STdeconvolve - Additional features with
STdeconvolve - Annotating deconvolved cell-types
- Analysis of 10X Visium data
- Examples of when
STdeconvolvemay fail - Discussion and examples of processing multiple datasets together or separately
Preprocessing datasets
For commands to reproduce the preprocessing of certain datasets used in the manuscript, check out:
https://jef.works/STdeconvolve/
and scroll down to the section: Reproducing Analyses.